2D Contour plot in Matlab
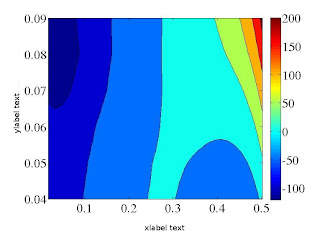
2D CONTOUR PLOT IN MATLAB %This .m file will generate 2D contour plot clear all; close all; L1 = load('dataset1.dat'); % [Let say the data set has 220 X co-ordinate data, and 6 Y co-ordinate data, and for these X and Y coordinate combinations Z coordinate values are there. Now, one has to form a matrix to get contour plot which will be generated in the following way] N1=6; N2=220; k = 1; for i = 1 : 1 : N1 for j = 1 : 1: N2 k = (i - 1) * N2 + j; X1(j,i) = L1(k, 1); Y1(j,i) = L1(k, 2); Z1(j,i) = L1(k, 3); end end contourf(X1, Y1, Z1); lighting gouraud xlim([0.0 0.6]) ylim([0.04 0.09]) set(gca, 'xtick', 0.0:0.1:0.6,'Fontsize', 15) set(gca, 'ytick', 0.04:0.01:0.09,'Fontsize', 15) xlabel('xlabel text','Fontsize', 20); ylabel('ylabel text','FontSize', 20);